Tumeurs oculaires : focus sur le mélanome choroïdien et le rétinoblastome

Mots-clés tumeurs, œil, mélanome, rétinoblastome, métastase
Copyright de toutes les images : Institut Curie
Résumé Le mélanome choroïdien est une tumeur intraoculaire, pigmentée et parfois achrome, se développant le plus souvent à l'âge adulte, et présentant un important risque métastatique. Le rétinoblastome est une tumeur de la petite enfance, responsable d'un reflet blanc dans la pupille, de diagnostic difficile car il survient à un âge pré-verbal. Leur traitement repose sur une prise en charge spécialisée afin de préserver en priorité la vie, l'œil et la vision.
Introduction Les tumeurs oculaires sont toutes des maladies rares, de diagnostic difficile et souvent tardif. Les lésions intraoculaires sont souvent détectées à un stade avancé, lorsque leur volume entraîne des signes visuels. Leur pronostic peut être sombre, la plupart étant des cancers agressifs, avec un potentiel métastatique. Leur prise en charge s'est codifiée au cours des dernières décennies avec la création de réseaux nationaux et européens, et de centres de références habilités à les diagnostiquer et à mettre en œuvre leur traitement. La prise en charge est pluridisciplinaire, et implique ophtalmologistes, radiothérapeutes, oncologues médicaux et onco-pédiatres, anatomopathologistes, radiologues et généticiens, ayant une expertise dans le domaine des tumeurs de l'œil. Les patients bénéficient de consultations d'annonce dédiées et de consultations infirmières de soutien lors du diagnostic, et les décisions sont prises en réunion de concertation pluridisciplinaire selon les bonnes pratiques en oncologie codifiées par l'Institut National du Cancer.
Dans cet article nous allons passer en revue les deux principales tumeurs intraoculaires primaires rencontrées en pratique clinique : le mélanome choroïdien chez l'adulte et le rétinoblastome chez l'enfant.
Mélanome choroïdien Diagnostic du mélanome choroïdien Il s'agit de la tumeur intraoculaire primitive la plus fréquente de l'adulte. Le mélanome uvéal peut toucher la choroïde (site le plus fréquent que nous décrirons ici), le corps ciliaire ou l'iris. Son incidence annuelle est d'environ 500-600 nouveaux cas par an en France. L'âge médian au diagnostic est de 62 ans mais il peut survenir à tout âge. Il touche généralement les sujets caucasiens, avec un surrisque parmi les personnes ayant les yeux bleus, pour des raisons non encore élucidées. Le diagnostic est clinique, ce qui est une singularité en oncologie, car l'obtention d'une preuve histologique par biopsie invasive exposerait à un risque de dissémination orbitaire. Il repose sur l'examen du fond d'œil et l'échographie oculaire. On observe une lésion choroïdienne en relief (Figures 1 et 2), de morphologie en dôme ou en champignon (Figure 3). La lésion est souvent pigmentée mais peut également être achrome (Figures 4 et 5), ce qui doit alors faire suspecter une métastase choroïdienne, avec réalisation d'un bilan d'extension complet, à la recherche d'une néoplasie primaire ayant métastasé à l'œil. Un décollement de rétine exsudatif peut être présent (Figure 6). Une hémorragie intravitréenne peut s'associer et masquer la tumeur. Ces deux présentations impliquent de devoir réaliser systématiquement une échographie oculaire en cas de décollement de rétine exsudatif sans déhiscence retrouvée, ou en cas d'hémorragie intravitréenne. En effet, une éventuelle chirurgie intraoculaire par vitrectomie sur un mélanome uvéal méconnu peut entraîner une dissémination tumorale.
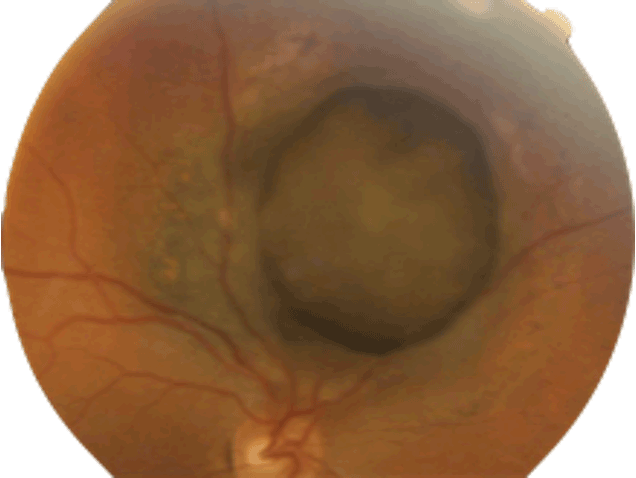
Figure 1 Mélanome choroïdien situé à proximité de l'émergence intraoculaire du nerf optique.
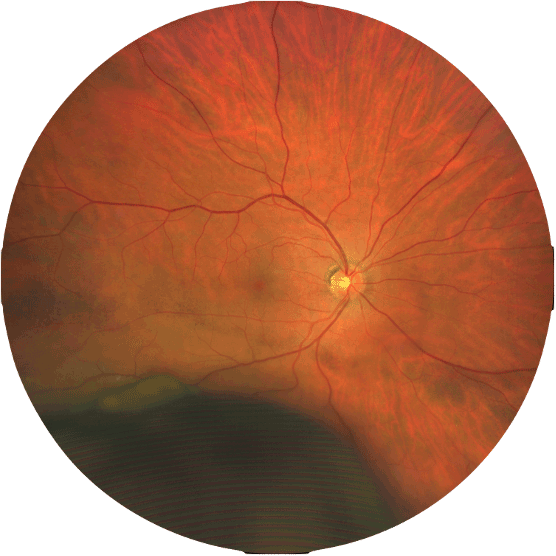
Figure 2 Mélanome choroïdien inférieur.
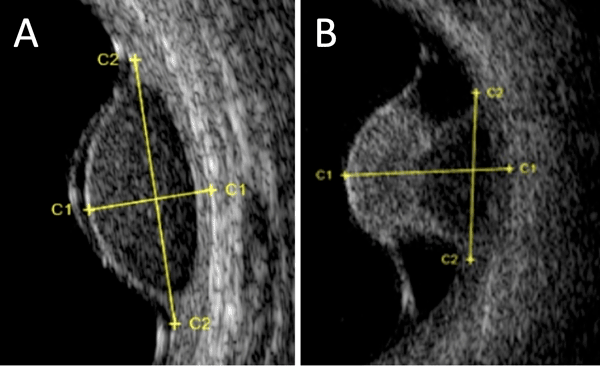
Figure 3 Échographie oculaire montrant un mélanome choroïdien en dôme (A) et en champignon (B).
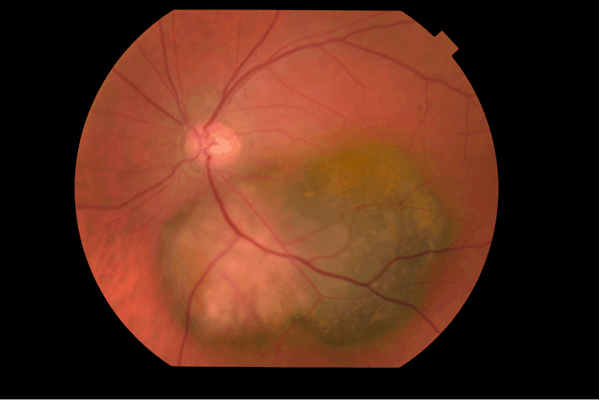
Figure 4 Mélanome choroïdien partiellement achrome.
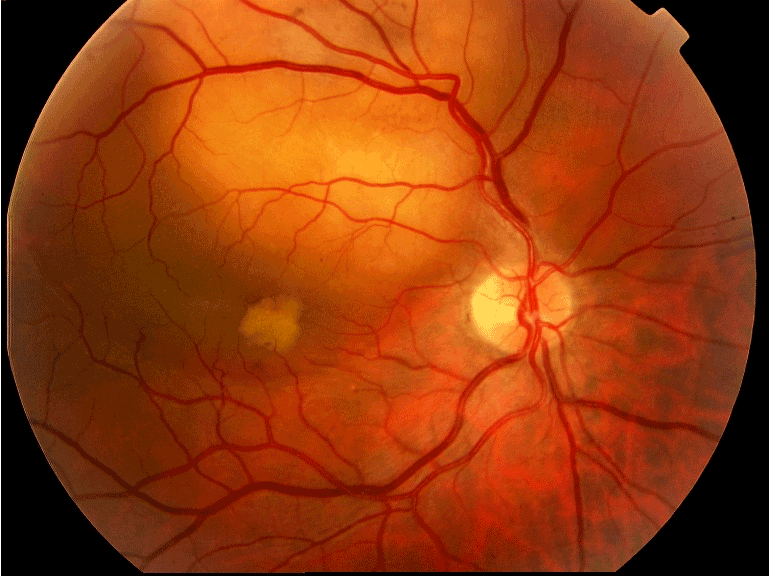
Figure 5 Mélanome choroïdien achrome.

Figure 6 Mélanome choroïdien avec des hémorragies de surface, associé à un décollement de rétine exsudatif sédimenté en inférieur. L'aspect strié des lésions provient d'un artéfact du système d'acquisition grand champ en raison du relief des lésions.
Traitement du mélanome choroïdien Les tumeurs d'épaisseur inférieure à 10 mm sont accessibles à une irradiation par protonthérapie dans un centre dédié (en France, les centres de protonthérapie d'Orsay et de Nice) ou par curiethérapie. Ces deux procédures nécessitent une chirurgie, soit pour positionner des clips de repérage à la surface de la sclère en vue d'une protonthérapie, soit pour suturer transitoirement un disque d'iode radioactif à la surface de la sclère. Dans les deux cas, la tumeur sera visualisée de façon indirecte par transillumination grâce à son épaisseur et sa pigmentation. Le contrôle tumoral après traitement par radiothérapie est de 96 % à 5 ans. Les tumeurs les plus volumineuses (épaisseur supérieure à 10 mm), ne peuvent pas bénéficier d'une irradiation en raison des effets secondaires oculaires qui seraient incontrôlables, notamment l'ischémie rétinienne induite et le syndrome de lyse tumorale qui entraineraient un glaucome néovasculaire (Figure 7). Une chirurgie d'énucléation est donc nécessaire, avec mise en place d'un implant orbitaire puis, dans un second temps, équipement par une prothèse calquée sur l'œil controlatéral, avec de très bons résultats esthétiques. Le globe oculaire sera analysé en anatomopathologie : en cas d'envahissement extra-oculaire, une radiothérapie orbitaire adjuvante sera réalisée.
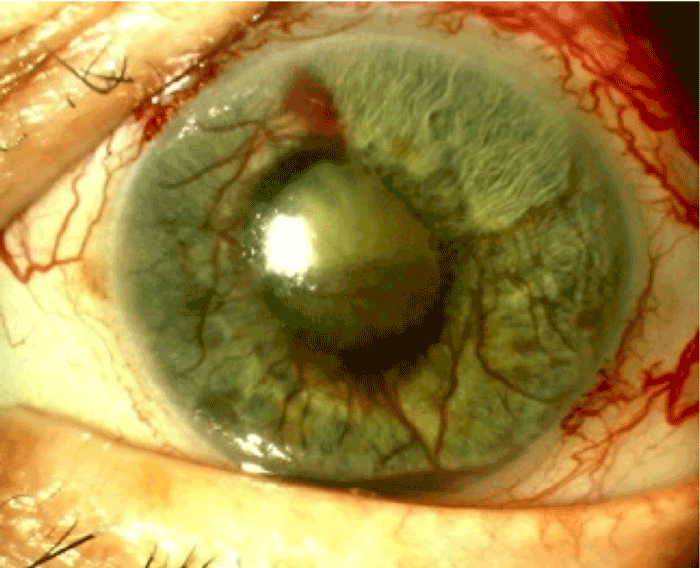
Figure 7 Glaucome néovasculaire compliquant un mélanome choroïdien de grand volume, par lyse tumorale et rétinopathie radiques responsables d'une lyse rétinienne.
Maladie métastatique Jusqu'à 50 % des patients atteints de mélanome uvéal développeront des métastases, principalement hépatiques (Figure 8). Les situations les plus à risque sont les tumeurs de grande taille et la génomique tumorale à haut risque (perte partielle ou totale d'un chromosome 3 et gain partiel du chromosome 8). Le risque génomique est estimé sur les pièces d'énucléation ou par ponction trans-sclérale lors de la pose des clips. Les patients effectuent une imagerie hépatique par échographie ou IRM semestrielle pendant 10 ans après le traitement initial. Si des lésions sont détectées, une exérèse chirurgicale des métastases localisées, ou un traitement par immunothérapie/thérapie ciblée/chimiothérapie pour des lésions multiples peut être proposé. Récemment, l'arrivée d'une nouvelle molécule, le tebentafusp, un anticorps bispécifique stimulant la réaction immunitaire contre les mélanocytes, a amélioré la survie globale, mais seuls les 45 % des patients sont éligibles, les porteurs d'un variant HLA précis (le HLA-A*02:01).
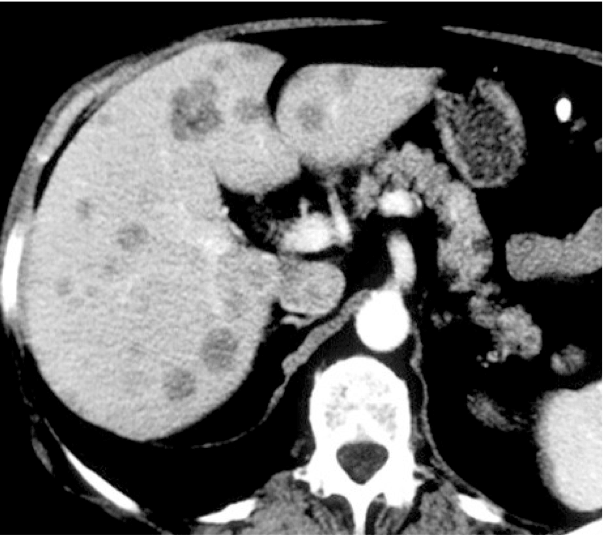
Figure 8 Scanner abdominal montrant de multiples métastases hépatiques d'un mélanome choroïdien.
Diagnostics différentiels du mélanome choroïdien
Le mélanome choroïdien peut être évoqué devant toute masse du fond d'œil, pigmentée ou achrome. Les diagnostics différentiels les plus souvent rencontrés sont :
- L'hémangiome choroïdien, lésion unique de tonalité plutôt rose-orangée, et responsable également d'un décollement de rétine exsudatif (Figure 9).
- Les métastases choroïdiennes, déjà évoquées plus haut, qui peuvent être confondues avec un mélanome achrome. Les métastases peuvent être multiples ou bilatérales, contrairement au mélanome. Elles s'accompagnent aussi d'un décollement exsudatif. Un bilan d'extension à la recherche d'une lésion primitive est essentiel (Figure 10).
- Les hématomes choroïdiens ou sous-rétiniens, souvent causés par des néovaisseaux méconnus (liés à une dégénérescence maculaire liée à l'âge par exemple) et favorisés par la prise d'anticoagulants ou d'anti-agrégants. Une surveillance régulière permet d'observer une résorption progressive de la lésion, contrairement à une tumeur qui grossirait progressivement (Figure 11).
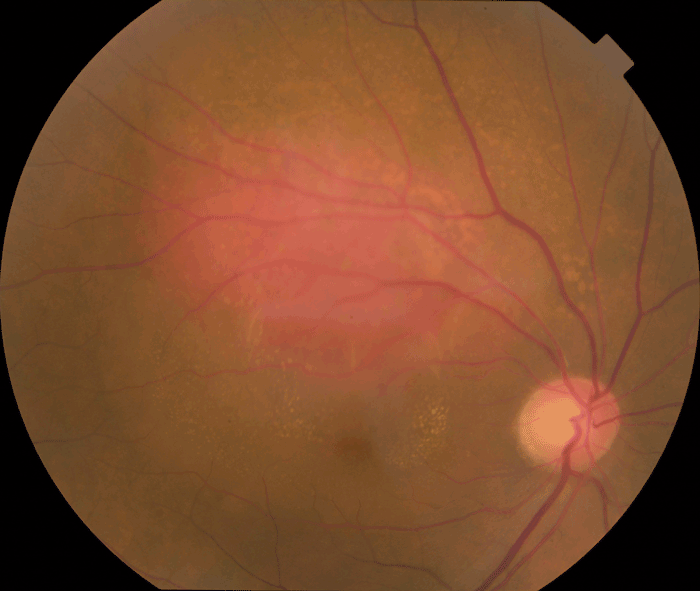
Figure 9 Hémangiome choroïdien se manifestant comme une masse rose-orangée.
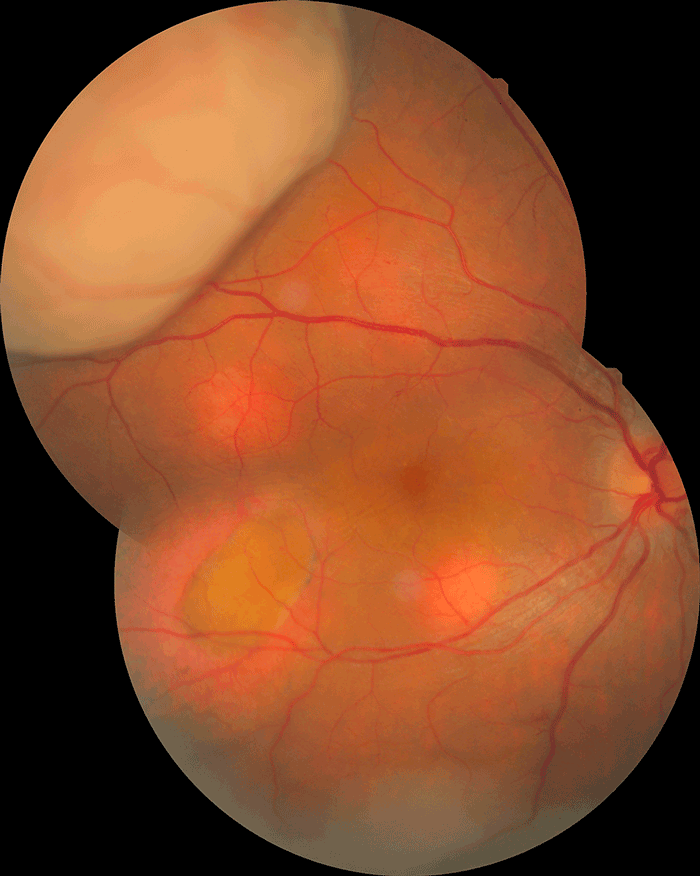
Figure 10 Métastases choroïdiennes multifocales d'un adénocarcinome pulmonaire.
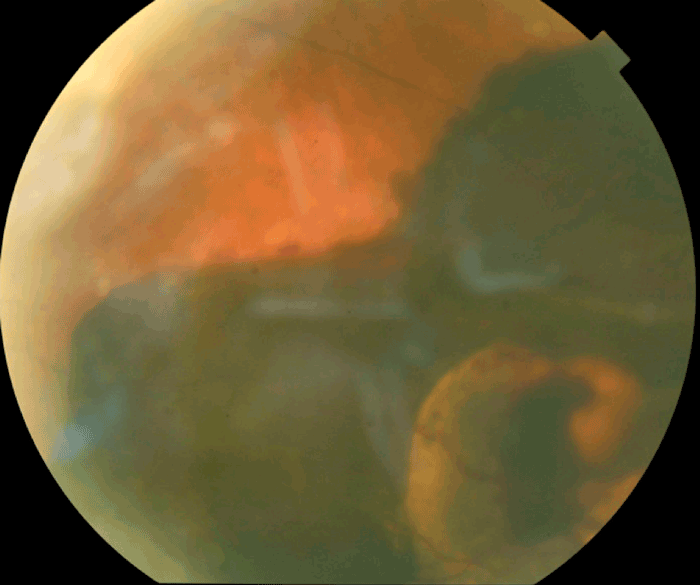
Figure 11 Hématome choroïdien compliquant une dégénérescence maculaire liée à l'âge néovasculaire.
Rétinoblastome Diagnostic du rétinoblastome Le rétinoblastome est une tumeur intraoculaire maligne de l'enfant dont on recense environ 50-60 nouveaux cas par an en France. L'âge moyen au diagnostic est de 1 à 2 ans selon les formes cliniques. C'est une maladie génétique, liée à la présence de mutations au niveau des deux allèles du gène RB1. L'atteinte peut être uni- ou bilatérale et un contexte familial est retrouvé dans 10 % des cas. L'atteinte est plus souvent bilatérale, multifocale, plus sévère et plus précoce en cas de prédisposition génétique. Le pronostic vital est excellent dans les pays à haut revenus, avec en France une survie supérieure à 99 %. Le pronostic visuel est cependant souvent moins favorable. Les signes d'appel les plus fréquents sont une leucocorie (Figures 12 et 13) et un strabisme. Une leucocorie est un reflet blanc au travers de la pupille parfois transitoire ou visible sur photo. L'évocation par les parents d'une leucocorie ou l'apparition d'un strabisme doit faire évoquer un rétinoblastome, et un fond d'œil doit être réalisé en urgence sous anesthésie générale. Au fond d'œil, on observe une ou plusieurs masses tumorales rétiniennes blanchâtres (Figures 14 et 15), parfois calcifiées, de dimensions variables, pouvant aller d'une lésion inframillimétrique débutante (Figure 16) à une tumeur volumineuse occupant toute la cavité vitréenne (Figure 17). Un décollement de rétine exsudatif, un essaimage tumoral sous-rétinien, intra-vitréen ou en chambre antérieure (Figure 18) peuvent être présents et sont de mauvais pronostic. Une IRM orbitaire doit être réalisée rapidement, pour confirmer le diagnostic et vérifier l'absence d'extension extraoculaire (Figure 19). Le scanner est déconseillé car l'irradiation est mutagène et augmente le risque de tumeurs secondaires en cas de prédisposition génétique. Les enfants en bas âge dans la fratrie devront également bénéficier d'un examen ophtalmologique complet, au besoin sous anesthésie.
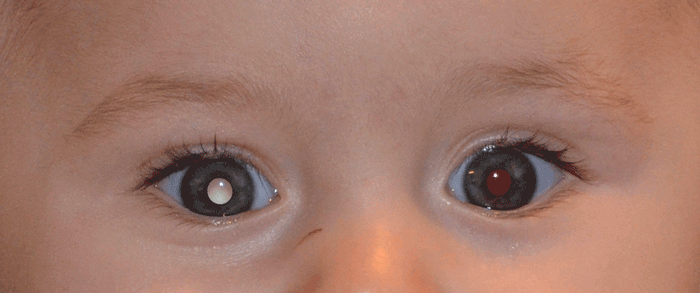
Figure 12 Leucocorie (reflet pupillaire blanc) causée par un rétinoblastome.
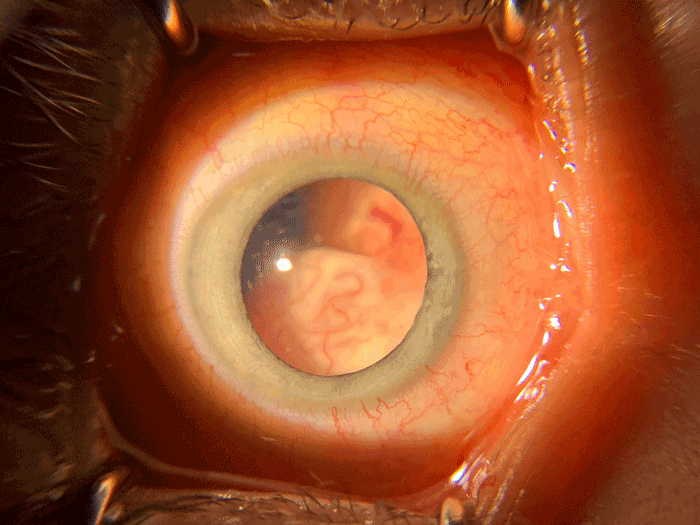
Figure 13 Masse intraoculaire blanchâtre, vascularisée, expliquant la leucocorie.
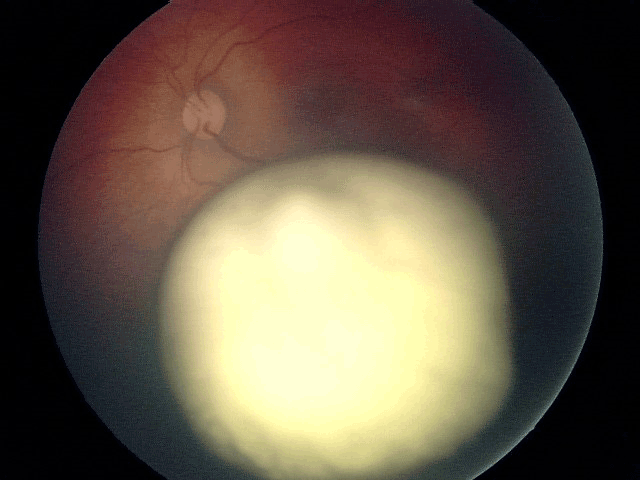
Figure 14 Masse blanchâtre en relief au fond d'œil correspondant à un rétinoblastome.
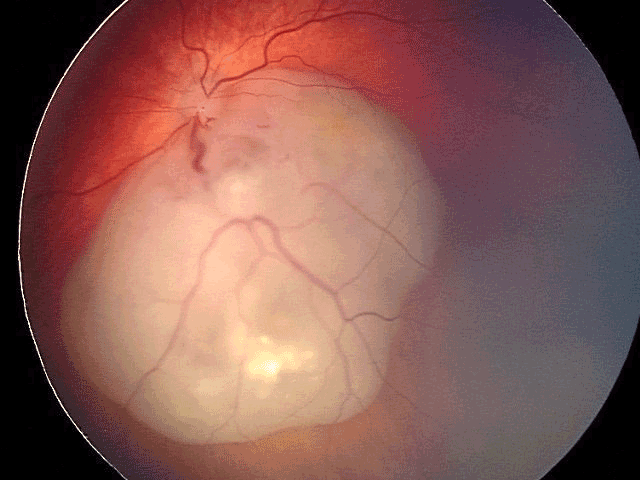
Figure 15 Masse blanchâtre au fond d'œil correspondant à un rétinoblastome. On observe quelques calcifications intratumorales caractéristiques.
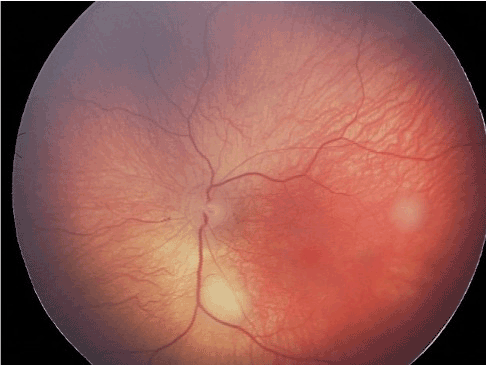
Figure 16 Petites lésions inframillimétriques correspondant à un rétinoblastome débutant.
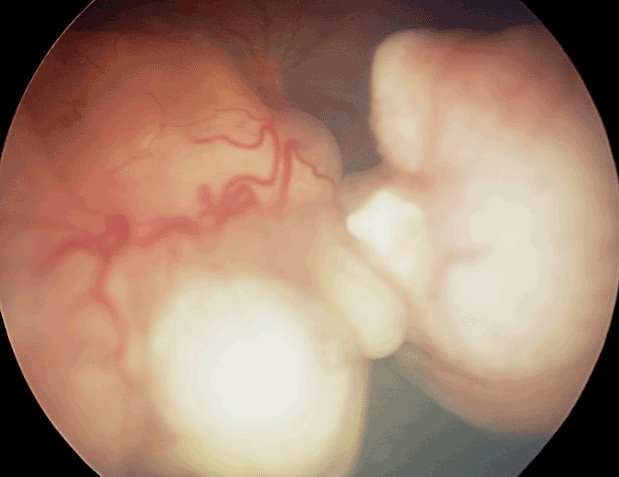
Figure 17 Volumineux rétinoblastome diagnostiqué à un stade avancé.
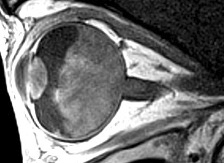
Figure 18 Essaimage tumoral dans la chambre antérieure de l'œil d'un rétinoblastome très avancé.
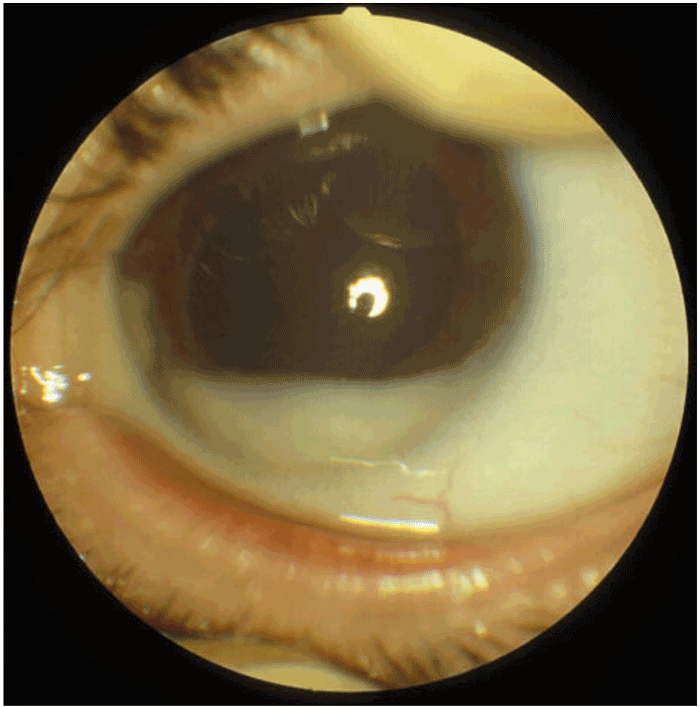
Figure 19 IRM montrant un rétinoblastome sans extension extraoculaire.
Traitement du rétinoblastome Le traitement dépend du type de rétinoblastome (uni/bilatéral), de la taille de la lésion et de l'invasion de structures comme le nerf optique, la chambre antérieure ou l'espace extrascléral. Un traitement par chimiothérapie sera administré par voie intra-veineuse ou par cathétérisme de l'artère ophtalmique depuis un abord fémoral, en lien avec un service de radiologie interventionnelle pédiatrique. Des traitements ophtalmologiques locaux (thermothérapie laser, cryothérapie, chimiothérapie intravitréenne) devront être associés, afin d'obtenir la régression de la tumeur dont il ne restera que des calcifications cicatricielles (Figure 20). En dernière ligne, on aura recours à une curiethérapie. Les tumeurs trop volumineuses ou étendues, en particulier avec extension extrasclérale, devront être traitées par énucléation, éventuellement complétée par une chimiothérapie systémique en cas de facteurs de risque histologiques de rechute métastatique. Enfin, dans les rares cas d'atteinte orbitaire, notamment en cas de retard diagnostic, une radiothérapie orbitaire pourra être nécessaire.
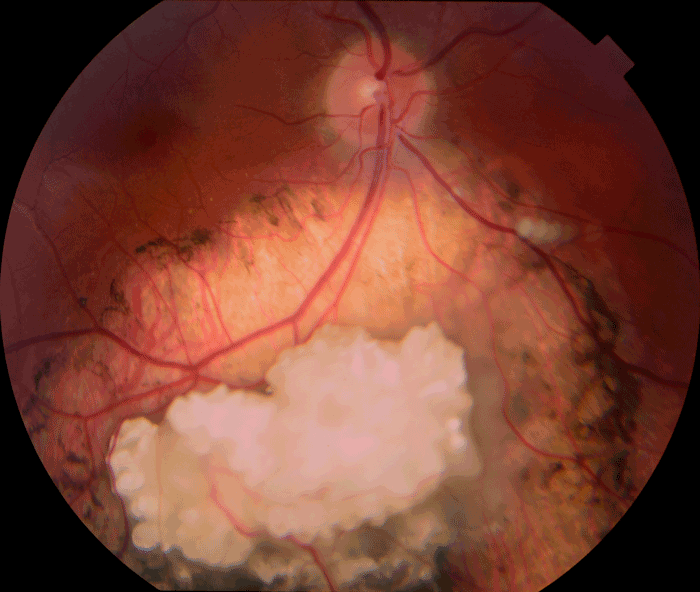
Figure 20 Rétinoblastome traité prenant l'aspect d'une cicatrice atrophique avec calcifications cicatricielles.
Diagnostics différentiels du rétinoblastome
Parmi les diagnostics différentiels les plus fréquents pouvant simuler un rétinoblastome, on peut citer :
- astrocytomes, petites lésions blanchâtres rétiniennes devant faire rechercher une sclérose tubéreuse de Bourneville (Figure 21).
- maladie de Coats : pathologie vasculaire responsable d'une exsudation lipidique très importante pouvant décoller la rétine (Figure 22).
- toute autre cause de leucocorie, comme par exemple une cataracte congénitale (Figure 23) ou un foyer infectieux rétinien (toxocarose, toxoplasmose, etc.).
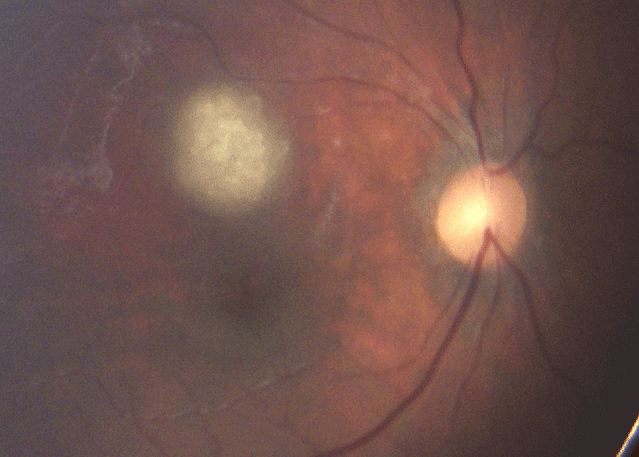
Figure 21 Astrocytome rétinien.
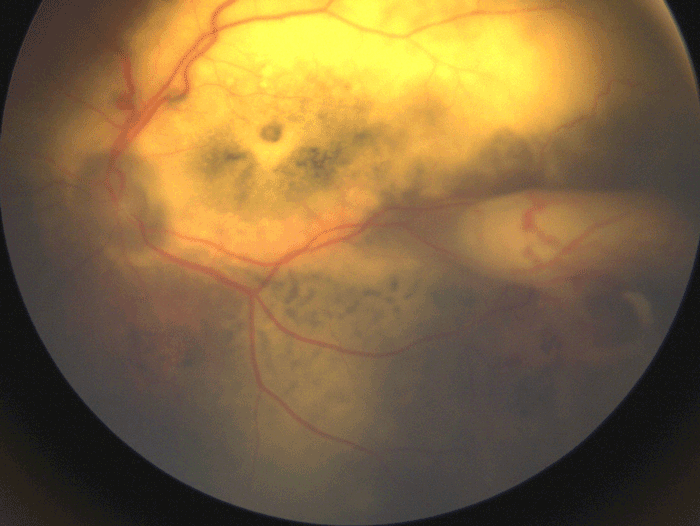
Figure 22 Maladie de Coats, maladie vasculaire rétinienne de l'enfant responsable d'une importante exsudation lipidique sous-rétinienne prenant un aspect « pseudo-tumoral » et pouvant être confondu avec un rétinoblastome.
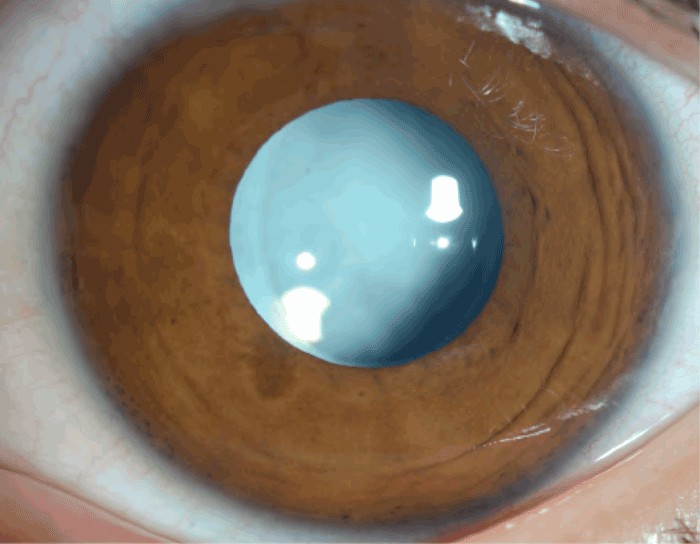
Figure 23 Cataracte congénitale, responsable d'une leucocorie de l'enfant.