Leucémie Lymphoïde Chronique : État de l'art

Introduction, diagnostic La leucémie lymphoïde chronique est une hémopathie maligne B considérée comme la leucémie la plus fréquente en Occident, avec en France plus de 4 000 cas par an, et environ 191 000 cas dans le monde[1,2]. L'âge médian du diagnostic est de 72 ans avec une prédominance masculine et un sex-ratio de 2:1. Il existe un sur risque (x 8.5) de développer une LLC chez les descendants d'un patient atteint de LLC et un sur risque chez les patients atteints de LLC de développer un cancer solide[3]. L'immunophénotypage permet de faire le diagnostic mettant en évidence une prolifération B mature CD5/CD19 avec plus de 5 000 lymphocytes B clonaux /mm3 pendant plus de 3 mois. Le diagnostic immunophénotypique repose sur le score dit RMH (voir tableau) qui affirme la LLC s'il est > 3 ; le myélogramme et/ou la biopsie médullaire sont inutiles au diagnostic dans ce cas[4].
Cotation 1 0 CD5 + - CD23 + - Expression slg monotypique Faible Forte FMC7 - + Expression de CD79b/CD22 Faible Forte
Une entité plus rare, le lymphome lymphocytique est en fait une prolifération initialement strictement ganglionnaire d'une population identique à celle de la LLC et dont l'évolution, le pronostic et le traitement ne sont pas différent de la LLC.
Classification et facteurs pronostiques La classification utilisée est celle de JL Binet datant de 1981[5] et est toujours utilisée différentiant 3 stades A, B et C. Les patients éligibles à un traitement étant les stade B évolutifs et stade C (iwcll).
Stade Aires lymphoïdes palpables Hémoglobine A Non B ≥ 3 Non C Indifférent Oui
Les principaux facteurs pronostiques actuellement reconnus sont les suivants[6] : - Sur le plan cytogénétique, la Del(17p), la Del(11q) sont considérées comme des facteurs défavorables, la Del(13q) lorsque unique comme favorable, la Del(17p) étant le facteur le plus important. - Un caryotype complexe (plus de 3 mutations clonales) ou hyper complexe (plus de 5 mutations clonales) est considéré comme défavorable. - Sur le plan moléculaire, la recherche d'une mutation ou d'une délétion de p53 ainsi que le statut du gène des immunoglobulines (IGHV) sont devenus des examens systématiques dont l'impact pronostique est important. Les facteurs défavorables sont un statut IGHV non muté et/ou un la présence d'une délétion/mutation de p53. - Sur le plan biochimique, le dosage de la beta 2 microglobuline (dont la valeur n'existe qu'en dehors de l'insuffisance rénale) et des LDH sont également des facteurs pronostiques importants.
Quels sont les dernières innovations en matière de facteurs pronostiques ? Le statut du gène p53 et du gène des immunoglobulines sont devenus la base du raisonnement pour orienter la prise en charge des patients. D'autres mutations ont été identifiées dont l'applicabilité en thérapeutique est pour l'instant modeste.
Bases de la prise en charge L'une des particularités de la LLC parmi les maladies malignes est qu'une majorité des patients (près de 80 %) ne nécessitent pas d'emblée de traitement de leur hémopathie car étant au stade A ou au stade B non évolutif de leur maladie ; cette attitude dite « watch and wait » a été documenté à la fin des années 1980 par le groupe français et les essais menés depuis, y compris les plus récents ont confirmé l'absence d'intérêt à une prise en charge préemptive7. Le suivi de ces patients doit se faire en fonction de la clinique, du temps de doublement des lymphocytes et en général de façon semestrielle. Il n'y pas d'indication à des examens radiologiques en dehors d'un point d'appel clinique.
Quels sont les critères d'initiation du traitement ?
Ils ont été définis en 2008 et actualisés en 2018[8] :
- Une atteinte médullaire progressive avec développement ou aggravation d'une anémie et/ou d'une thrombopénie. Les seuils habituellement considérés pour l'initiation d'un traitement spécifique sont un taux d'hémoglobine < 100 g/l ou un taux de plaquettes < 109/l. Néanmoins, certains patients considérés comme en stade C de Binet du fait d'une thrombopénie modérée peuvent rester stables et asymptomatiques pendant une longue période sans traitement. Dans ce cas, l'évolution des cytopénies au cours du temps est à prendre en compte avant de décider de la mise en route d'un traitement.
- Une splénomégalie massive ou symptomatique ou augmentant de taille progressivement.
- La présence de ganglions volumineux ou symptomatiques ou augmentant de taille progressivement.
- Une lymphocytose progressive, avec augmentation de plus 50 % sur une période de deux mois ou un temps de doublement des lymphocytes (TDL) inférieur à 6 mois. Ce critère ne doit pas être utilisé chez les patients avec un chiffre initial de lymphocytes < 30 x 109/L, et doit être interprété dans le contexte clinique global.
- Une anémie et/ou thrombopénie auto-immune répondant insuffisamment aux corticostéroïdes ou aux autres traitements usuels.
- La présence de signes généraux définis par l'un ou plus des signes ou symptômes ci-dessous en rapport avec la maladie :
- Perte de poids de 10 % ou plus non volontaire dans les 6 mois précédents,
- Asthénie significative (ECOG PS 2 ou plus ; incapacité à effectuer les activités usuelles,
- Fièvre de plus de 38.0°C durant 2 semaines ou plus sans signe d'infection,
- Sueurs nocturnes d'une durée de plus d'un mois sans signe d'infection.
Quels sont les examens recommandés avant d'initier un traitement ? Ils sont résumés dans le tableau ci-dessous.
obligatoire recommandé Beta 2 microglobuline x Caryotype x FISH 4 sondes Del 11q Del13q Tri12 del17p x x x x Statut mutationnel IGHV x Mutation TP53 x TDM TAP x
À noter que la recherche pas NGS ciblé (NOTCH, SF3B1, BIRC3, ATM…) n'est recommandée que dans le cadre des essais cliniques.
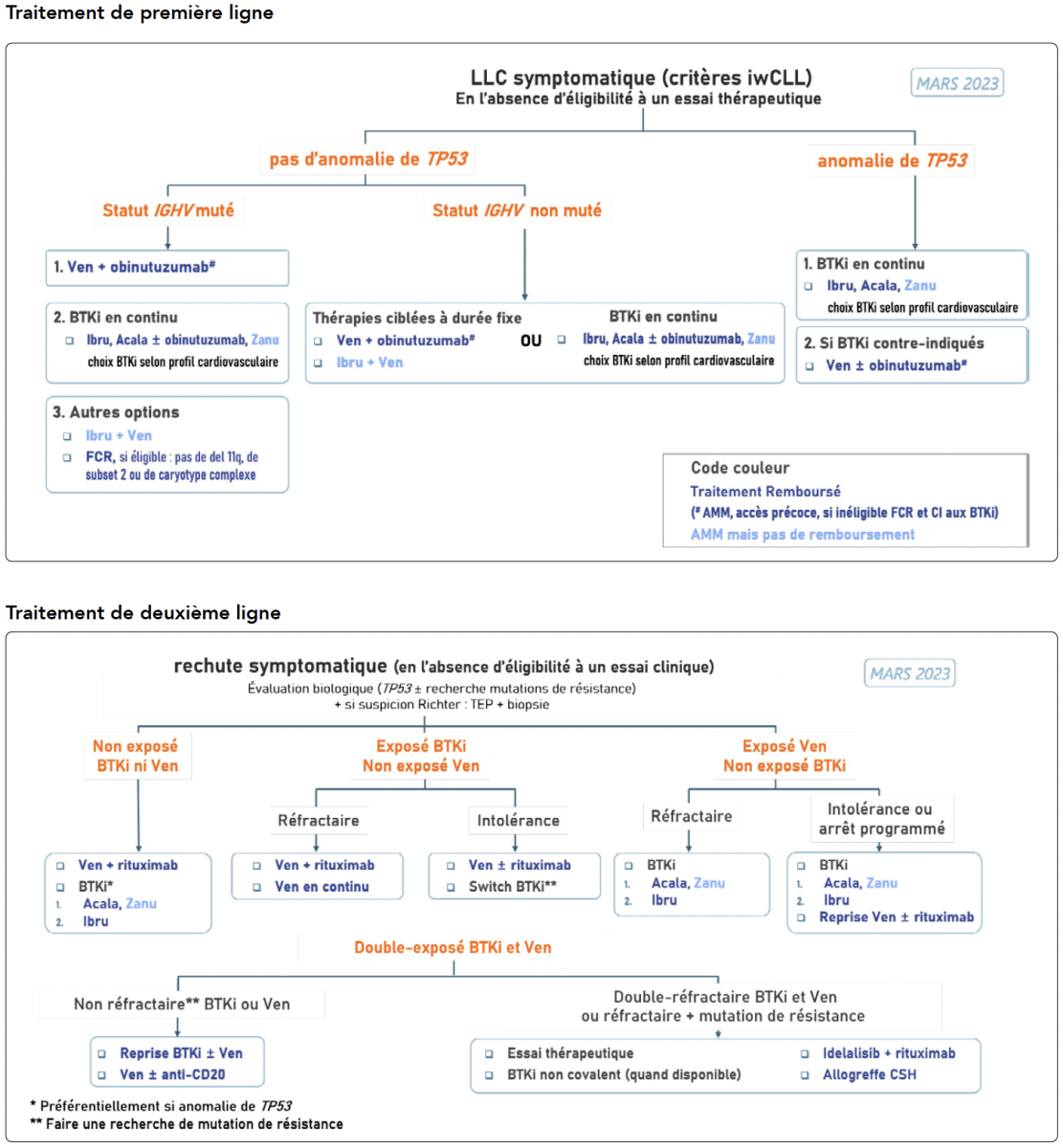
Traitement En moins de dix ans le traitement de la LLC a basculé de l'immunochimiothérapie de type fludarabine-endoxan-rituximab (FCR) ou bendamustine- rituximab (BR) ou chlorambucil- rituximab (ou obinutuzumab) à des traitements dits ciblés. Deux grandes classes médicamenteuses sont actuellement disponibles : (i) les inhibiteurs de BTK (iBTK) ibrutinib, acalabrutinib, zanubritinib, qui sont prescrit jusqu'à progression ou toxicité et les inhibiteurs de BCL2 (venetoclax) qui est prescrit un an en première ligne et 2 ans en deuxième ligne. La deuxième génération des iBTK (acalabrutinib, zanubrutinib) semblent être grevée d'une moindre toxicité (saignements, troubles du rythme, HTA) avec une efficacité non inférieure. Les inhibiteurs de BCL2 sont plus contraignants à l'initiation du traitement en raison d'une période de ramp-up rendue nécessaire par le risque de syndrome de lyse, mais sont particulièrement bien tolérés par la suite. Les stratégies proposées par le groupe National FILO sont résumées dans les 2 tableaux suivants, ne laissant qu'une place très marginale à l'immunochimiothérapie. Ces stratégies reposent sur 2 examens biologiques, le statut TP53 et IGHV, les choix étant également fonction des co-morbidités et co-médications des patients. Pour une revue récente des essais publiés voir référence 9.
LLC et risque infectieux La prévention des infections est un enjeu majeur dans la LLC, tous les patients étant à risque même si non traités.
Vaccination[10] Les vaccinations doivent être à jour : (i) vaccination annuelle contre la grippe, (ii) vaccination anti-pneumococcique : PREVENAR 13 suivi, au moins 8 semaines plus tard de PNEUMOVAX avec rappel vaccinal par PNEUMOVAX proposé à 5 ans, (iii) vaccination anti-Haemophilus, (iv) vaccination contre le SARS-COV-2 selon les recommandations du moment. Les vaccins vivants sont contre-indiqués.
Antibioprophylaxie[11] Celle-ci n'est pas systématique et les recommandations sont variables en fonction des pays et des modalités de traitement. Les modalités précises de prise en charge sortent du cadre de cet article.
Supplémentation en immunoglobulines polyvalentes (IgIV)[12] L'hypogammaglobulinémie est quasi-constante dans la LLC et va en s'aggravant avec l'évolution de la maladie. La substitution en immunoglobulines est indiquée, après avis en RCP, en cas de dosage des Ig G< 4g/L associé à des infections à répétition ou entraînant une hospitalisation.
Les complications auto-immunes Elles sont fréquentes, touchant jusqu'à 15 % des patients et posent d'importants problèmes de prise en charge. Elles sont largement dominées par les anémies hémolytiques auto-immunes (+++) les thrombopénies et érythroblastopénies. En cas de survenue en dehors d'une évolutivité de la LLC sous-jacente, le traitement de première ligne reste la corticothérapie à 1-2 mg/kg qui permet d'obtenir 70 % de réponse rarement durables. Les IgIV sont peu efficaces et réservées à l'urgence. Les analogues de la thrombopoïétine peuvent avoir un intérêt. Le rituximab est une option en 2e ligne comme la ciclosporine. En cas de cytopénie réfractaire, un traitement de la LLC doit être proposé. En cas de survenue dans un contexte de LLC évolutive la prise en charge est complexe et repose en général sur une corticothérapie initiale associée à l'endoxan et le rituximab (RCD). La place des traitements ciblés est difficile à définir et se décide au coup par coup. La prévention des infections opportunistes est dans tous ces cas importante, surtout si un inhibiteur de BTK est utilisé. Le traitement de l'érythroblastopénie est complexe et repose sur la ciclosporine, le rituximab, le RCD, l'ibrutinib et/ou les IgIV en fonction des situations. Pour une revue générale voir référence 13.
Le lymphome de Richter Il s'agit d'une complication évolutive de la LLC, caractérisée par une transformation en lymphome B diffus à grande cellules agressif. La présentation est souvent bruyante et il s'agit dans 80 % d'une maladie clonalement reliée à la LLC. De rares cas de Richter sont d'authentiques maladies de Hodgkin. Le pronostic de la maladie reste très mauvais, avec des médianes de survie de 15 à 27 mois et de faibles taux de réponse complète. Le traitement conventionnel est le même que celui du lymphome (type R-CHOP) ou des traitements associant les sels de platine à la cytarabine et, pour l'instant, l'adjonction ou la substitution des nouvelles molécules n'a pas apporté de bénéfice significatif. Pour une revue récente voir référence 14.
Conclusion La LLC a vu sa prise en charge radicalement modifiée en moins de sept ans et son pronostic significativement s'améliorer avec l'utilisation des traitements ciblés et des anticorps anti-CD20. La maladie reste cependant à ce jour non curable, et il persiste de nombreux champs où d'importants progrès sont nécessaires tels la prise en charge du déficit immunitaire, des complications dysimmunitaires et du lymphome de Richter.